Multiple cerebral cavernous malformations in association with a Dubowitz-like syndrome
Article information

Abstract
Cerebral cavernous malformations (CCMs) are proliferative sinusoidal vascular lesions and are the most common vascular malformations of the brain. They can occur sporadically or secondary to an underlying genetic predisposition where multiple lesions are commonly seen. Dubowitz syndrome is a clinically-diagnosed rare genetic disorder with an unknown molecular basis. An association between these conditions has not been reported previously. A 30-year-old woman with a Dubowitz-like syndrome presented with acute left leg weakness, gait ataxia and transient loss of consciousness. Imaging revealed five CCMs with recent hemorrhage in relation to one lesion in the left middle cerebellar peduncle. A recurrent hemorrhage from the same lesion occurred ten weeks later and she underwent microsurgical excision of this malformation. Genetic analysis revealed an unbalanced chromosomal rearrangement involving partial deletion of chromosome 7q21, the locus of the CCM1/KRIT1 gene known to be associated with familial CCMs. This is the first description of CCMs in association with the Dubowitz phenotype. The genetic basis of Dubowitz syndrome may be heterogeneous but, for the first time, overlap is demonstrated between this condition and multiple CCMs, with a possible common genetic etiology. Knowledge of this association may be of help in the management of acute neurological presentations in Dubowitz-like syndromes.
Keywords: Hemangioma, Cavernous, Central nervous system, Dubowitz syndrome, Genetics
INTRODUCTION
Cerebral cavernous malformation (CCM) – also known as cavernous (haem)angioma or cavernoma – is the most common vascular malformation of the brain with a lifetime incidence of 0.4–0.8%. Up to 40% are diagnosed incidentally, a proportion which is likely increasing due to the widespread availability of magnetic resonance imaging (MRI).1)17) CCMs are hamartomatous lesions of thin-walled, endothelium-lined, sinusoidal vascular chambers without intervening brain tissue, often likened in macroscopic appearance to a mulberry. They occur either sporadically or as the consequence of a familial or genetic predisposition where multiple lesions are most commonly encountered. Here we report a case of multiple CCMs diagnosed in an adult female with a clinical diagnosis of Dubowitz syndrome, a rare genetic disorder of multiple congenital abnormalities, growth retardation, intellectual disability and other multisystemic features. The results of chromosomal analysis are presented and reveal a putative common etiology for the syndrome and the multiple CCM disorder. To our knowledge, an association between these conditions has not been described previously.
CASE REPORT
A 30-year-old woman was admitted as an emergency to our neurosurgical unit following a collapse with transient loss of consciousness lasting a few minutes, which was immediately preceded by acute left leg weakness and vertigo. At the time of initial assessment there was residual subjective left lower limb weakness and no other neurological symptoms.
Her past medical history included a childhood diagnosis of Dubowitz syndrome, associated intellectual disability, moderately excessive alcohol consumption, eczema, cataract and multiple terminations of pregnancy. She had attended a special needs school until the age of fifteen and had achieved some secondary school qualifications. She was self-caring, lived in her own residence with 24 hours assistance from on-site support workers, and was not in employment. Family history included a niece also diagnosed with Dubowitz syndrome but was otherwise negative for vascular or neurological disease.
On examination, she was alert and orientated. Microcephaly, retrognathia, short stature, eczematous skin and a high-pitched yet hoarse voice in keeping with underlying Dubowitz syndrome were evident.15) There was mild weakness of the left lower limb of Medical Research Council (MRC) grade 4, with normal tone and symmetrical reflexes. She was independently ambulant with a broad-based gait but was unable to perform tandem walking unassisted. Apart from a long-standing partial right ptosis there were no other focal neurological deficits.
Brain computer tomography revealed multiple hyperattenuating supra- and infra-tentorial intraparenchymal lesions. The initial working diagnosis was intracerebral metastases but was revised after MRI of the brain was performed. MRI studies confirmed the presence of five intra-axial lesions: one in the left middle cerebellar peduncle and one each in the right frontal, right parietal, right temporal and left frontal lobes. Imaging characteristics were in keeping with multiple CCMs (Fig. 1) with features of recent hemorrhage in relation to the left middle cerebellar peduncle lesion. The final diagnosis was left middle cerebellar peduncle hemorrhage on a background of multiple CCMs. The patient underwent physical rehabilitation and was discharged home with outpatient follow-up and a plan to refer her to another neurosurgical centre for consideration of elective stereotactic radiosurgery.

brain MRI of the five cerebral cavernous malformations (CCMs) at initial presentation. Four of the lesions were apparent on T1-weighted (T1w) and T2-weighted (T2w) images: right frontal (A), right parietal (B), right temporal (C) and left middle cerebellar peduncle (D). All lesions are intraparenchymal (marked with arrows) and show the typical ‘pop corn’ appearance of CCMs with heterogeneous signal internally, surrounded by a T1w and T2w hypointense rim of haemosiderin. The right frontal and parietal lesions are shown on susceptibility weighted imaging (SWI) to demonstrate the characteristic blooming artefact (E, left image). The fifth, small left frontal lesion was apparent only as a focus of signal drop-out on SWI (E, right image). MRI, magnetic resonance imaging.
Ten weeks later she was re-admitted with acute headache, nausea and vomiting, associated with deteriorating mobility. She was now unable to ambulate independently due to marked gait ataxia. There was MRC grade 4 weakness and incoordination of the left upper and lower limbs, and horizontal nystagmus in all directions of gaze. Repeat imaging showed acute hemorrhage with increased mass effect at the site of the left middle cerebellar peduncle malformation (Fig. 2).
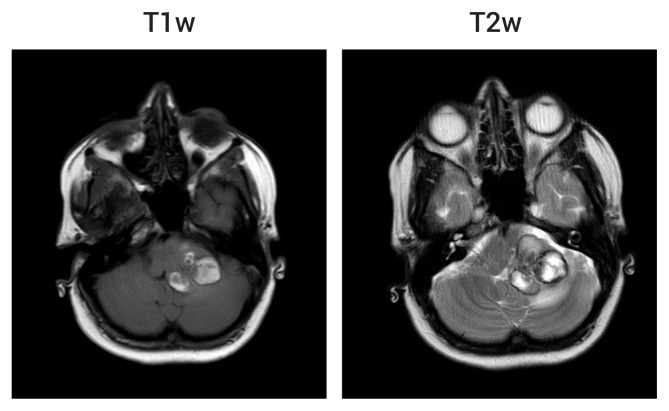
brain magnetic resonance imaging following the second clinical presentation. There are new areas of T1w hyperintensity and T2w iso- to hypo-intensity at the site of the left middle cerebellar peduncle cerebral cavernous malformation suggestive of acute hemorrhage. There is an associated increase in mass effect on the surrounding brainstem and cerebellum when compared to the previous images (Fig. 1D).
Surgical indications included recurrent symptomatic hemorrhage within three months and the increase in mass effect on the brainstem and we decided to perform expedited microsurgical excision of the left middle cerebellar peduncle CCM via a retrosigmoid approach. Following surgery she recovered well with mild residual left-sided limb ataxia and was discharged home on the seventh post-operative day after a short period of rehabilitation. Histopathological analysis of the excised lesion confirmed the diagnosis of CCM.
In view of the multiplicity of lesions and her developmental background, she was referred to the regional clinical genetics service for further evaluation. Chromosomal analysis identified an abnormal maternally-inherited chromosome 7 with an unbalanced translocation believed to be cause of her underlying Dubowitz-like phenotype. This comprises insertion of chromosome 3q23.1-q26.2 into chromosome 7q, coupled with deletion of 7q21.11-q21.2 at the insertion site. Thus, there is a single-copy partial deletion of 7q21, a locus including the CCM1/KRIT1 gene implicated in familial multiple CCM syndromes.11)
DISCUSSION
CCMs most commonly arise sporadically but can occur as a consequence of a familial or genetic disorder in at least 6% of cases.1)8) Genetic forms of CCM are characterized by the presence of multiple lesions but more than one malformation may also be found in 12–20% of sporadic cases. Symptomatic lesions commonly present with seizures when supratentorial, focal neurological deficit usually when infratentorial, or headache.1)17)
Three genes have been implicated in the pathogenesis of familial CCM: CCM1 (KRIT1), CCM2 (MGC4607) and CCM3 (PDCD10).2)3)5)7)9)11) These are believed to account for the majority of genetic cases and are inherited in an autosomal dominant manner with incomplete penetrance. The molecular pathogenesis of CCMs remains unknown but is believed to follow a Knudsonian ‘two-hit’ mechanism.8)
Dubowitz syndrome is a rare condition of presumed but unknown genetic cause first described in 1965.6) By 1996 only 141 cases had been described in the literature.15) It has been described as a syndrome characterized by multiple congenital abnormalities, intellectual disability, growth failure, immune defect predisposing to allergies and eczema, with a propensity in some cases to develop blood dyscrasias, haematologic malignancy and neuroblastoma.15) It is phenotypically variable and diagnosis is made clinically on the basis of a number of common features, as evident in the subject of this report, including: characteristic facies with retrognathia, large low set ears, ptosis and prominent nasal bridge; microcephaly; intrauterine growth retardation and short stature; mild intellectual disability; and eczema. It has been suggested that the condition may be a group of genetically heterogeneous disorders with similar phenotypic presentation.12)16) Structural neurological abnormalities are uncommon, however individual cases of ventriculomegaly, cerebral atrophy, myelomeningocoele, neuronal heterotopia, midline dysgenesis of the corpus callosum and pituitary, and bony abnormalities of the craniocervical junction have been described.10)13)14) Cerebrovascular manifestations are extremely rare with only one case of complete internal carotid artery occlusion and one of multiple saccular aneurysms reported in the literature, though in the latter the diagnosis of Dubowitz syndrome was questioned.4)15)
We believe that this is the first description of CCMs in a patient with clinical features of Dubowitz syndrome. Lesion multiplicity strongly pointed to an underlying genetic cause and chromosomal analysis subsequently revealed an unbalanced rearrangement involving partial deletion of chromosome 7q21, a locus harbouring the CCM1/KRIT1 gene. Dubowitz syndrome probably encompasses a genetically heterogeneous group of conditions with overlapping phenotypic presentation.12) Some putative causative genetic abnormalities have been described including microdeletions/duplications of parts of chromosomes 13q, 14q and 17q, as well as mutations in the genes NSUN2 (chromosome 5p), LIG4 (chromosome 13q) and PCNT (chromosome 21q).16) We believe that this is the first time that a Dubowitz-like phenotype has been associated with a chromosome 7q21 abnormality and with multiple CCMs through a potential common genetic mechanism incorporating deletion of the CCM1/KRIT1 gene at this locus.
CONCLUSIONS
We report a case of symptomatic multiple cerebral cavernous malformations (CCMs) in a 30-year-old woman with features of Dubowitz syndrome. This is, to our knowledge, the first description of CCMs within the clinical spectrum of Dubowitz-like syndromes, with a putative common genetic etiology involving chromosome 7q21 and the CCM1/KRIT1 gene. Awareness of this association may help in the future management of patients with Dubowitz-like syndromes presenting to clinical neuroscience centres with acute neurological disturbance and may shed light on the genetic basis of this condition.
Notes
Disclosure
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.